
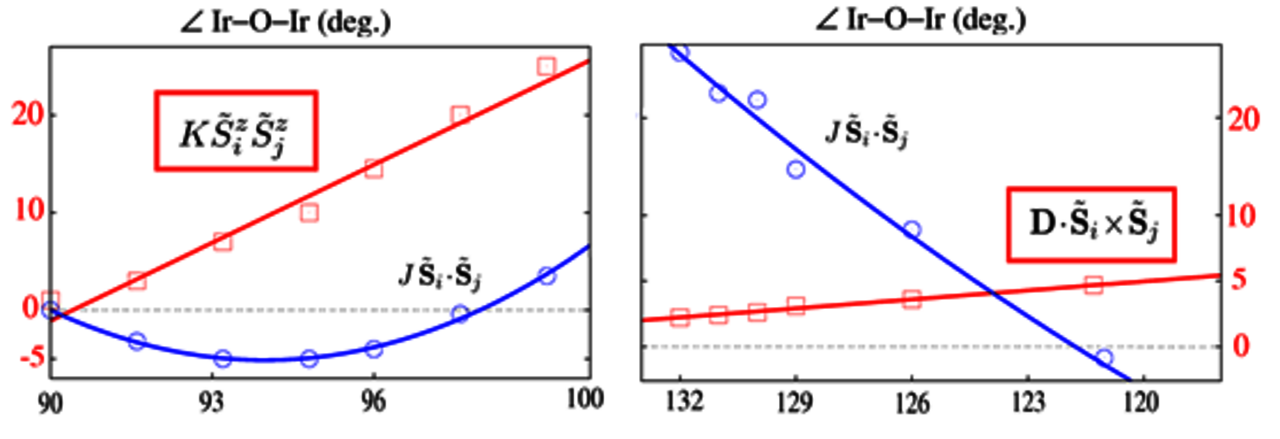
The quantum chemistry team uses ab initio wave-function-based methods to understand the electronic structures and various properties of materials. Special attention is given to transition-metal and more recently 4f compounds, with focus on the interplay of electronic correlations, spin-orbit couplings, and structural effects. By performing the calculations at levels of increasing sophistication (Hartree-Fock, multiconfiguration self-consistent-field, multireference configuration-interaction etc.), the role of different types of correlation effects (d-d correlations, ligand-to-metal charge-transfer-type correlations etc.) in relation to particular material characteristics can be transparently disentangled. This refers to d- and f-shell multiplet structures and excitation energies, g factors, effective intersite spin couplings, band gaps and band widths etc. Such studies provide valuable insights into the essential mechanisms behind various properties, allowing on one hand a correct interpretation of available experimental data and secondly predictions for material trends.
| Pritam Bhattacharyya | p.bhattacharyya(at)ifw-dresden.de |
| Mohamed S Eldeeb | m.s.eldeeb(at)ifw-dresden.de |
| Liviu Hozoi | l.hozoi(at)ifw-dresden.de |
| Thorben Petersen | t.petersen(at)ifw-dresden.de |
| Name | Dr. Liviu Hozoi |
| Address | IFW Dresden Helmholtzstraße 20 01069 Dresden |
| Phone Number | +49-351-4659-1829 |
| l.hozoi(at)ifw-dresden.de |


